VAE molecule generation
Replication of A Two-Step Graph Convolutional Decoder for Molecule Generation paper by Bresson et Laurent (2019)

Introduction
In this post, I discuss how I replicated the model and results from the 2019 ML paper: A Two-Step Graph Convolutional Decoder for Molecule Generation.
Literature read
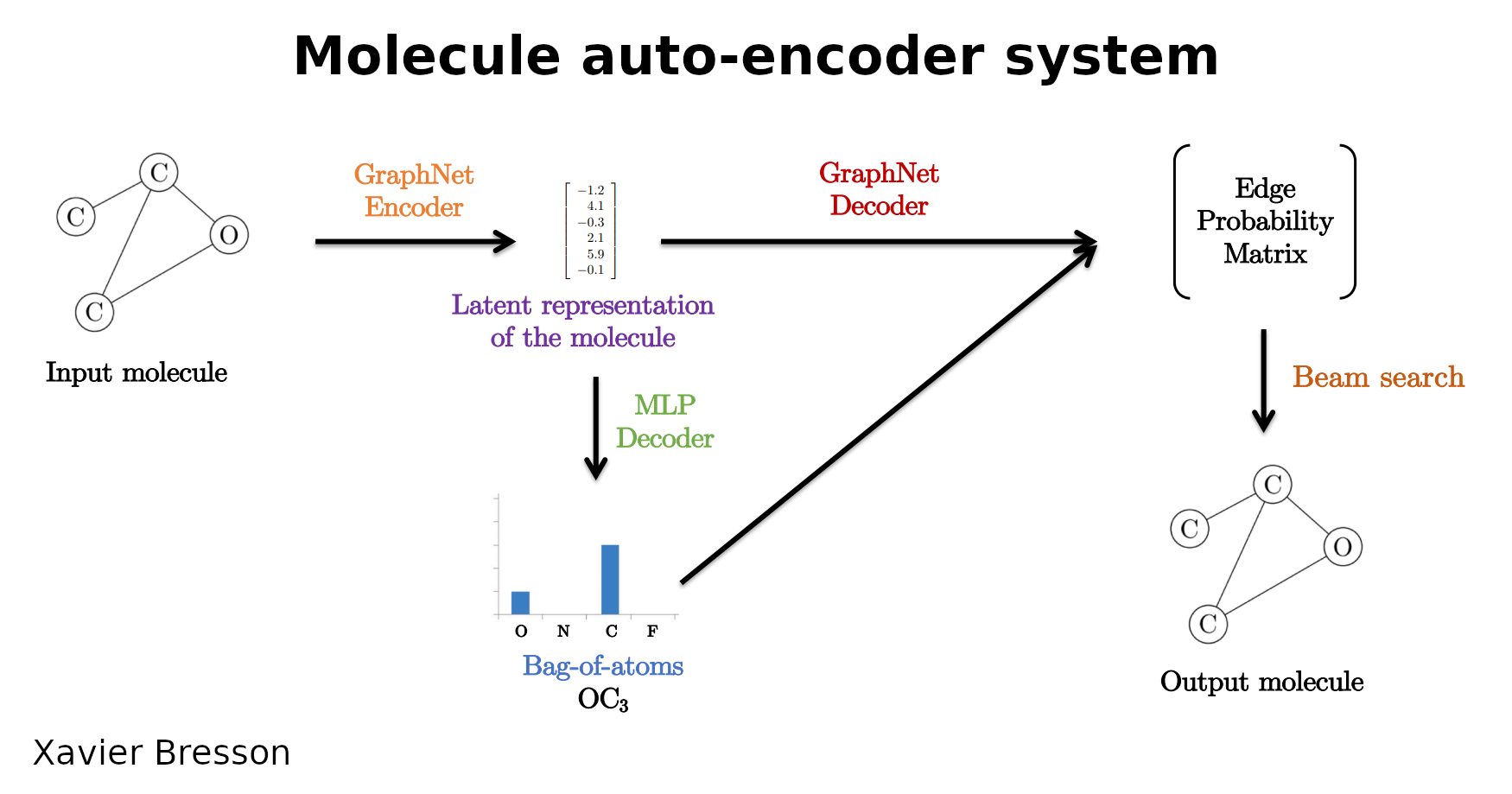
I started by reading the paper. As illustrated in the flowchart above, the authors use the following approach to train the ML model:
- Given an input SMILES string, convert it to a numerical graph representation (vertices are atoms, edges are bonds)
- Train a graph convolutional network on the graph (more specifically, a Residual Gated Graph ConvNet). This step then generates a latent representation of the molecule (graph) which is a single summary vector $z$
- Using $z$, train a neural network classifier to determine the number of atoms for each predicted atom type (e.g. 3 carbon and 1 oxygen atom). The authors refer to this as a ‘bag-of-atoms’ matrix
- Using the bag-of-atoms matrix, create a fully connected graph such that all atoms (vertices) are connected to each other
- Together with $z$, the bag-of-atoms matrix, and this fully connected graph, train another Graph ConvNet to generate a bond (edge) level feature prediction
- Determine loss from ground truth bag-of-atoms and actual edge features (single bond, double bond, etc.)
One additional constraint is that latent representation should be Gaussian. Or more formally, $z$ represents the distribution of the latent space which should be as close to Gaussian as possible. This formulation is known as a Variational Autoencoder and it allows us to sample from a unit Gaussian distribution to generate molecules. For inference, the authors use the following approach:
- Sample from a unit Gaussian distribution to generate the latent representation $z$
- Generate the bag-of-atoms matrix and the bond features similar to step 3-5 above
There are additional considerations such as positional encoding and edge probability loss calculations which will be covered in the following sections. The paper also discusses additional post-generative steps including beam search and molecule validity checks, but I ignored those for simplicity.
Tools and libraries
To replicate the paper, the following components were used:
- Python as the program language
- Pandas for processing the molecular dataset
- RDKit for molecule specific functions such as SMILES conversion
- PyTorch for creating the model, training loop, and inference pipeline
- PyTorch Geometric for the graph-specific ML algorithms and data batching
- Streamlit for visualization
As a first step to building the system, I accessed and explored the training dataset.
Dataset exploration
The authors used a subset of the ZINC dataset. Since I was already using PyTorch Geometric, I thought I could simply load in the ZINC dataset which is bundled with the library. However, this method doesn’t allow us to extract any SMILES or molecule information directly since the atom and bond feature mappings are not explicitly shared. Instead, I did my own processing on raw SMILES string with a dataset I found on Kaggle: ZINC-250k dataset. For debugging, I loaded a small subset of 1000 molecules.
You can see how the data and relevant features were processed with the following widget:
I’m actively working on the rest of the post! Check back soon…
References
A huge thanks to the following authors for writing introductory tutorials on Variational Autoencoders: